
Drs. Carl Maki and Lei Duan Lab Group
Carl Maki, PhD, is a professor in the Department of Anatomy & Cell Biology. He earned his bachelor’s degree in biology in 1987 from the University of Wisconsin Lacrosse and his PhD in 1993 from Kansas State University. Maki received post-doctoral training in cancer biology at the National Institutes of Health and Harvard Medical School. He has held faculty positions at the Harvard School of Public Health, the University of Chicago and Rush University.
Our research
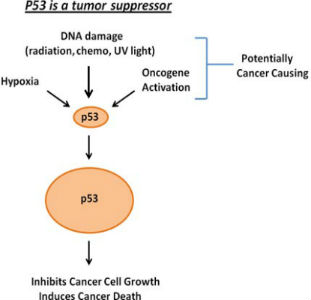
Dr. Maki works closely with Lei Duan, MD in the Department of Anatomy and Cell Biology and together they direct a cancer research lab. One focus of the lab is the p53 tumor suppressor. Wild-type p53 is a transcription factor and is expressed at low levels in most cells. However, p53 levels increase, and the protein is activated in response to DNA damage and other stresses. Activated p53 then inhibits growth or induces cell death.
In addition to activating genes p53 can also repress transcription through a transcription repressor complex called DREAM. P53 represses expression of different mitosis promoting genes through the DREAM complex, and we showed this repression improves cancer therapy responses and contributes to tumor suppression. We also showed p53 activity is regulated by a protein called CSE1L.
Duan L, Perez RE, Calhoun S, Maki CG. RBL2/DREAM-mediated repression of the Aurora kinase A/B pathway determines therapy responsiveness and outcome in p53 WT NSCLC. Sci Rep. 2022 Jan 20;12(1):1049.
Duan L, Tadi MJ, Maki CG. CSE1L is a negative regulator of the RB-DREAM pathway in p53 wild-type NSCLC and can be targeted using an HDAC1/2 inhibitor. Sci Rep. 2023;27;13(1):16271.

In this picture human lung cancer cells were untreated (top right) or treated with Nutlin-3a (bottom right) to activate wild-type p53. Cytoskeleton proteins were visualized by immunostaining. The results show that Nutlin-3a alters the cancer cell cytoskeleton. This resulted in decreased ability of these cancer cells to migrate. See this publication: “Nutlin-3a Induces Cytoskeletal Rearrangement and Inhibits the Migration and Invasion Capacity of p53 Wild-Type Cancer Cells.”
Moran DM, Maki CG. Mol Cancer Ther. 2010 Apr;9(4):895-905.
Current Studies
Mutations in p53 are the common genetic alteration in cancer, occurring in over 50% of cases. In one our projects, the Maki-Duan group identified a drug combination that can effectively target cancer cells and has selectivity against cancers with p53 mutated or deleted. Current studies are aimed at testing this drug combination in vivo and determining the mechanism how it works.
TK is a protein that functions in mitosis and regulates the spindle assembly checkpoint. Small molecule inhibitors of TTK are current clinical trials against HER2-negative breast cancer. However, a challenge is that not all cancers respond to TTK inhibitors, and there are no molecular markers to identify cancers that will or will not respond. In one of our projects we identified a gene signature that appears capable of predicting sensitivity to TTK inhibitors in current clinical trials. Current studies are examining if this signature can predict tumor responsiveness to TTK inhibitors, and how the factors encoded by these genes affect sensitivity.
Current Funding

Department of Defense W81XWH2210134 (Maki)
Co-investigator (Duan)
Rational targeting of TTK/MPS1 in HER2-negative breast cancer
03/2022-02/2025
NIH NCI R21CA273658 (Maki)
Co-Investigator (Duan)
A synthetic lethal approach for targeting p53 deficient triple negative breast cancer
04/2023-03/2025
Swim Across America 2024
PI (Maki)
A novel therapeutic approach for triple negative breast cancer
Publications
A complete listing of Maki’s publication can be found on PubMed.
Contact
Carl Maki, PhD
Professor
Department of Anatomy & Cell Biology